
High-entropy alloy catalysts: high-throughput and machine learning-driven design

Abstract
High-entropy alloy (HEA) catalysts have recently attracted worldwide research interest due to their promising catalytic performance. Most current studies focus on designing HEA catalysts through trial-and-error methods. This produces scattered data and is not conducive to obtaining a fundamental understanding of the
Keywords
INTRODUCTION
The widespread prevalence of catalysts in industry determines human living standards, as more than 85% of chemicals are made through catalytic reactions[1-3]. In their inception phase, with the growing understanding of catalysis, a series of catalysts with high catalytic efficiency have been developed and designed, resulting in the rapid development of catalysis[4-6]. The development of catalysts has been mainly focused on the two strategies of simplification and complication. The former involves reducing the dimensions (i.e., 3D → 2D → 1D → 0D) or decreasing the particle size of the catalysts. Regarding complication, single-atom catalysts (SACs) take this strategy to the extreme and have become the most popular catalysts due to their high catalytic performance[7,8]. The large-scale production of SACs, however, still presents an enormous challenge. Researchers have attempted to synthesize complex morphologies or structures to improve catalytic performance. Alloying has been proven to be one of the most effective methods by various research groups through the design of different alloy systems, including binary, ternary and quaternary alloys, with excellent catalytic performance[9-12].
As opposed to SACs, which have the simplest active site, high-entropy alloy (HEA) catalysts have more complex active sites due to their large compositional space and diverse atomic arrangements[13]. HEAs are composed of five or more elemental components in near-equimolar ratios and were first reported by Cantor and Yeh in 2004[14,15]. Due to their rich compositional and configurational spaces, some novel HEAs with specific mechanical properties have been designed and synthesized by direct current magnetron
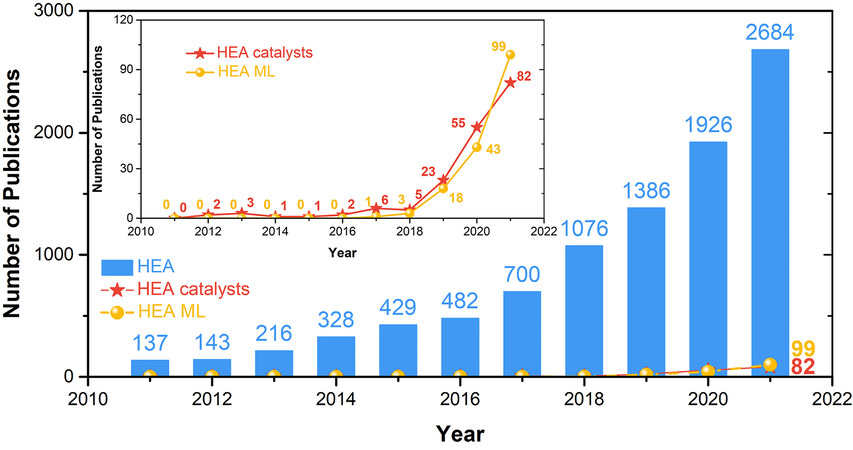
Figure 1. Number of publications on HEAs, HEA catalysts and HEA machine learning (ML) by 2021. The data are based on the Web of Science. HEA: high-entropy alloy.
Although HEAs have demonstrated excellent catalytic performance for various catalytic reactions, such as the HER, oxygen reduction reaction (ORR), oxygen evolution reaction (OER), CO2 reduction reaction (CO2RR), MOR and ammonia decomposition reaction, their structure-property-performance relationships are still ambiguous due to the complex active sites of HEA catalysts. A HEA with a face-centered cubic (FCC) structure containing five elements should have 510 = 9,765,625 active sites on its (111) surface when only the top site (one atom) and the nearest neighbor atoms (nine atoms) are considered active centers, while it should be 515 and 518-519 for bridge (two active atoms + 13 neighbor atoms) and hollow (hollow-FCC: three active atoms + 15 neighbor atoms; hollow-hexagonal-closed packed (HCP): three active atoms + 16 neighbor atoms) sites, respectively. Herein, symmetry was not considered for calculating the number of active sites since there is no symmetry on HEA surfaces when considering second nearest neighbor atoms. Such vast possibilities make it impossible for them to be investigated experimentally or computationally. Until now, the development of HEA catalysts has been sluggish and random, limited by the traditional “trial-and-error” methods or “directed research” approaches[25]. The available data are limited for understanding the nature of HEA catalysts. Consequently, the greatest challenge is identifying the active center in order to explore the structure-property-performance relationships of HEA catalysts. Only by fully understanding HEA catalysts can the rational design of HEA catalysts be implemented to make a breakthrough in catalysis.
To address the above challenge, more data are needed to analyze the structure-property-performance relationships of HEA catalysts. However, it is difficult to compile the single data points achieved by traditional “trial-and-error” methods and “directed research” approaches into an effective database due to the variance in synthesis methods, particle sizes, morphologies, and so on. Moreover, it is prohibitively time-consuming and costly to collect the data points one by one. To solve this issue, high-throughput (HT) techniques have been proposed to generate comprehensive databases with high efficiency[26]. With the enhancement of computing power and the continuous improvement of theoretical calculation methods, HT theoretical calculations play an essential role in building databases as a result of their fast speed and low cost when compared with HT experiments. This does not mean that HT experiments are unnecessary, as they are essential to building a realistic database. To better understand HEA catalysts, a large database is a prerequisite and rational analysis of these data is indispensable. The diversity of HEA catalysts, however, leads to complex databases, which are difficult to analyze using only physical and chemical knowledge.
Fortunately, machine learning (ML) has received significant attention in recent years as a powerful tool for processing complex data[27-30]. For example, to understand the structure-activity relationships of SACs for nitrogen fixation, different 2D materials supporting SACs and boron-doped graphene SACs were explored using HT density functional theory (DFT) calculations and ML[28,29]. Deng et al. applied DFT calculations and ML to develop bi-atom catalysts for the ORR[30]. These ML models provide new insights into atomic catalysts and help to speed up the design and discovery of new atomic catalysts. HEA catalysts are much more complex than atomic catalysts and the HT and ML methods show significant promise in exploring the structure-property-activity relationships of HEA catalysts, as evidenced by the works of Wan et al. and
In this review, we summarize the current HT techniques on the synthesis, characterization and performance testing used in the discovery of new HEA catalysts. Based on the achieved database, some ML models have been developed to analyze the structure-activity relationships and predict the catalytic activity of HEA catalysts for various catalytic reactions, such as the HER, OER/ORR, CO2RR and ammonia decomposition or synthesis. After an overview of the application of HT techniques and ML models in HEA catalysts, we focus on the challenges, opportunities and prospects in the development of HEA catalysts.
HT RESEARCH FOR HEAs
HT techniques are essential for scientists to efficiently generate large databases and the subsequent information extraction[33,34]. For HEAs with huge composition space, HT techniques can be used to great effect for the discovery and development of HEAs[35]. HT techniques make HEA research more automatic, parallel and efficient[36]. The application of HT techniques in the development of HEA catalysts is summarized from two aspects, namely, HT theoretical calculations and HT experimental approaches.
HT theoretical calculations
With the rapid development of high-performance computing and theoretical calculation methods, HT theoretical calculations have become more efficient in generating material databases compared to experiments[37]. Both DFT and semi-empirical calculations of phase diagrams (CALPHAD) have shown to be viable and popular approaches for investigating the atomistic and thermodynamic mechanisms of existing HEA systems, as well as for new HEA catalyst design[38].
The first step of a theoretical calculation is to build a HEA model, which is necessary for targeted and rapid HEA discovery and application[39]. Currently, the most widely used method to build HEA structures is the special quasi-random structure (SQS) generation approach, which combines the cluster expansion technique and Monte Carlo algorithms through several codes (ICET, ATAT, MCSQS and Supercell)[40-43]. The designed SQSs can model disordered alloys with atomic resolution and the radial distribution function of a random system is a quintessential concept for the generation of realistic random structures[44]. Moreover, by combining artificial neural networks (ANNs) and evolutionary algorithms, our group proposed a neural evolution structure (NES) generation methodology for HEA structure generation
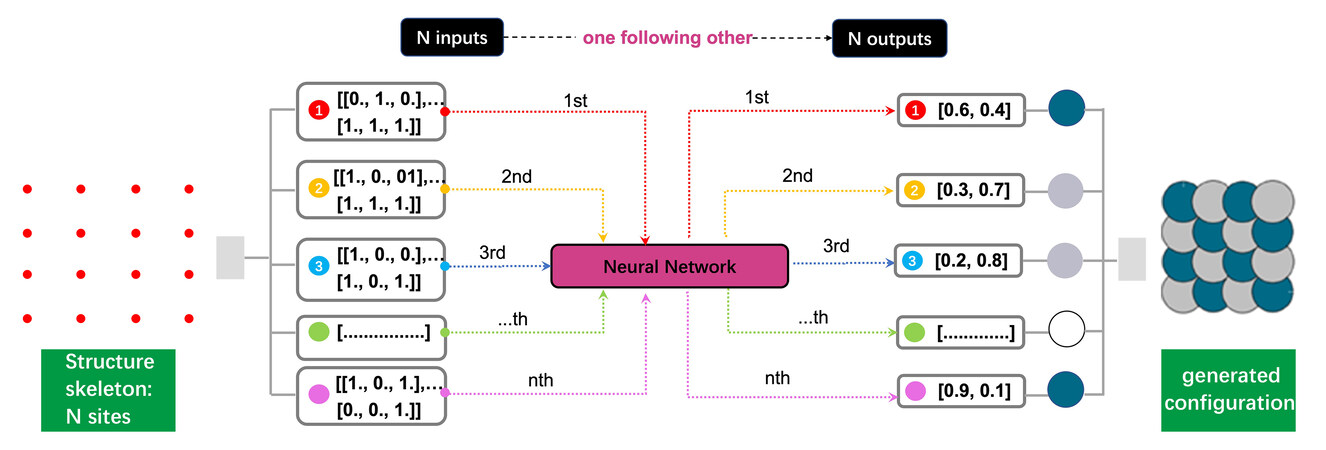
Figure 2. Roadmap of NES generation process. From left to right: an input structure (a template mesh with N sites), a representation of the N input arrays, the ANNs, a representation of the N output vectors, the atom type associated with each output vector and the generated configuration. Reproduced with permission[45]. Copyright 2021, AIP Publishing. NES: neural evolution structure.
Surface energies and work functions are directly related to adsorption performance and can therefore be used as descriptors of catalytic activity[28,46,47]. Duong et al. calculated the surface energies and work functions of Co-Cr-Fe-Mn-Mo-Ni alloys using HT DFT calculations[48]. In this work, low-order FCC alloys (up to quaternary) with different alloy compositions belonging to the senary Co-Cr-Fe-Mn-Mo-Ni system were considered in calculating their surface energies and work functions. The CALPHAD methodology was applied in the sub-regular solution model to analyze the populated first-principles data. CALPHAD models allow smooth interpolations across the alloy composition domains, as well as extrapolations to higher-order FCC HEAs, achieving more information with fewer data. In addition, considering the error of any models, Bayesian statistics were adopted to quantify, to some extent, the uncertainties of the CALPHAD models. Note that in this work, the achieved surface energies and work functions by HT calculations were used to rank the inherent corrosion-resistance potential of various equiatomic and near-equiatomic FCC HEAs belonging to the Co-Cr-Fe-Mn-Mo-Ni system, rather than their catalytic performance. We believe that HT calculations of surface energies and work functions show significant potential for the application of HEAs in catalysis because these surface properties are closely related to surface catalysis.
The adsorption energy of a specific intermediate has been considered as a descriptor of the catalytic performance for the corresponding catalytic reaction. This can be utilized to deliver prominent results due to the Brønsted-Evans-Polanyi (BEP) relationship between activation barriers and reaction energies and the scaling relationship among the adsorption energies of some intermediates on catalyst surfaces[49,50]. To reduce the calculation cost, it is necessary to only consider the adsorption energy of some important intermediates to evaluate the catalytic performance of the catalysts[49]. Taking the CO2RR as an example, various products can be produced at different potentials, while product distributions also depend on the solvent, promoter and other factors[51,52]. Roy et al. considered that the intermediates of CO*, HCO*, H2CO* and H3CO* are more important for the formation of methanol from CO2, even though several CO2RR mechanistic pathways are possible for methanol formation[53]. Moreover, the catalyst for the electrocatalytic CO2RR should not be active for the HER and oxidized during the reaction process. Thus, except for CO*, HCO*, H2CO* and H3CO*, the adsorption energies of H* and O* were calculated by HT DFT calculations. The neighboring atoms of the active center on a certain catalyst play a vital role in determining the adsorption energy of a given intermediate adsorbed on the active center. In this work, the neighboring atoms were divided into three regions based on their varying effects on the adsorption energy. The first region is the atoms on the adsorption site; the second region includes the surface layer atoms around the adsorption site and the third region contains the subsurface layer atoms, which are the nearest neighbors of the adsorption sites, as shown in Figure 3A-C. A database with 474 data points in more than 40,000 possible microstructures was generated for the CO2RR. Based on this database, the authors built an ML algorithm to predict the adsorption energies of the important intermediates on all possible adsorption sites. This work demonstrates an efficient approach to exploring catalysts with high catalytic performance, which can be extended to other reactions in the future.
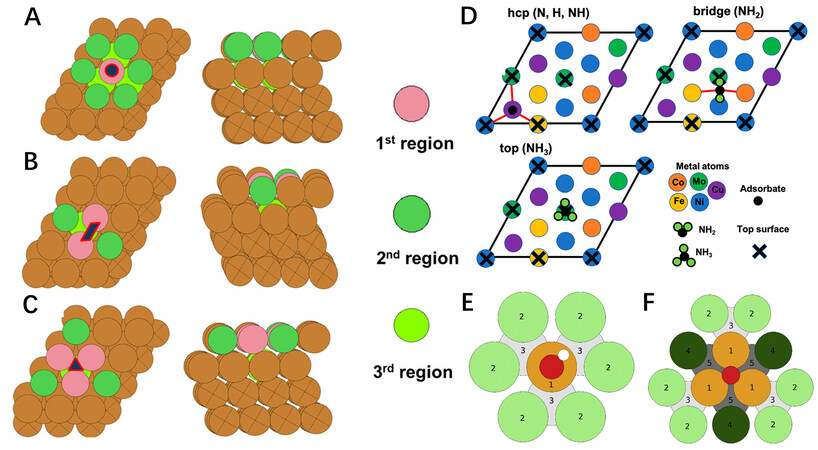
Figure 3. Top and side views of (A) on-top, (B) bridge and (C) hollow-HCP surface microstructures in a 3 × 4 × 4 slab, where atoms with different colors denote different regions. (D) Top view for adsorption configurations for different chemical species on a representative HEA FCC (111) surface. Atoms belonging to the top surface layer are labeled with a cross. Bonds between adsorbates and metal atoms are shown as red lines. Parameterization of the surface configurations is achieved using nearest neighbors. (E) OH* on-top binding. Each colored zone represents a set of three parameters. Orange (1): binding site. Light green (2): surface neighbors singly coordinated to binding site. Light gray (3): subsurface neighbors singly coordinated to binding site. (F) O* FCC hollow site binding. Each colored zone represents a set of five parameters, except for zone 1, where the set is 35 parameters. Zones 1-3 as in (E) and additionally dark green (4): surface neighbors doubly coordinated to binding site. Dark gray (5): subsurface neighbors doubly coordinated to binding site. (A-C) Reproduced with permission[53]. Copyright 2021, American Chemical Society. (D) Reproduced with permission[56]. Copyright 2021, American Chemical Society. (E) and (F) Reproduced with permission[60]. Copyright 2019, Elsevier. HCP: hexagonal-closed packed; FCC: face-centered cubic; HEA: high-entropy alloy.
For ammonia decomposition or synthesis, N*, NH*, NH2* and NH3* are important intermediates for adsorption energy calculations, as well as H*, since the HER is the most important side reaction for ammonia synthesis[54,55]. Saidi et al. performed HT DFT calculations to determine the adsorption energies of H*, N*, NH*, NH2* and NH3* species on FeCoNiCuMo HEA surfaces[56]. It was found that the most stable adsorption site changes from the HCP site to the bridge site and finally to the top site with increasing H atoms, as shown in Figure 3D. Based on these adsorption configurations, they established a database with 1911 configurations. Combined with data analytics and ML, the scaling relationships among the binding energies of NHx* (x = 0, 1, 2 or 3) can still be seen in FeCoNiCuMo, as in the case of monometallic surfaces[57,58]. These correlations indicate that the adsorption energy of N* could be a good descriptor for ammonia decomposition and synthesis on these HEAs.
The adsorption of OH* on Pt(111) is ~0.1 eV stronger than the optimum adsorption energy based on Sabatier volcano plots for catalyzing the ORR[59]. Batchelor et al. presented HT DFT calculated adsorption energies of OH* and O* on the (111) surface of an IrPdPtRhRu HEA[60]. As shown in Figure 3E and F, the most stable adsorption sites of OH* and O* are the top and FCC hollow adsorption sites, respectively. A model was established to link the local atomic arrangement around the adsorption sites to the adsorption energy, which can predict the adsorption energy values on all possible surface sites. Our group extended this model from the (111) surface to other Miller index surfaces, including the (100), (110), (211) and (532) of an equimolar FCC IrPdPtRhRu HEA[61]. A total of 12 types of coordination environments were considered for HT DFT calculations of the adsorption energy of OH* [Figure 4A], including more than 1,000 data points. According to these data points, we developed and trained a neural network model with high accuracy and universality. The ligand and coordination effects on the adsorption energy were analyzed sequentially by the feature importance based on our developed neural network model [Figure 4B]. Quantitatively, the adsorption energy was found to have a linear relationship with the total coordination number of nearest neighbors. More interestingly, the neural network model can be simplified to a simple linear scaling relationship with only a slight loss of accuracy. It has been demonstrated that these recently developed neural network techniques are powerful tools that can be used to overcome the huge chemical space of HEA catalysts.
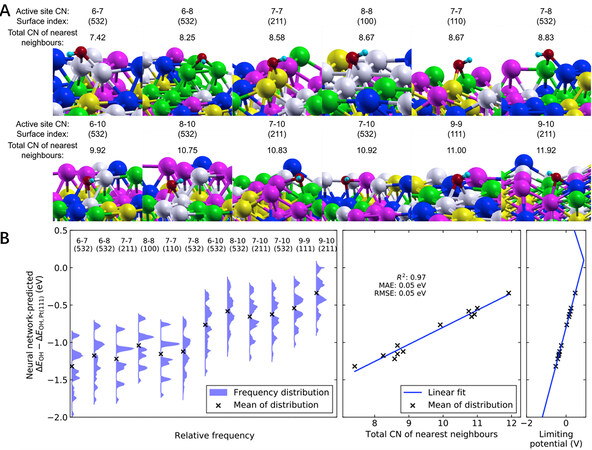
Figure 4. Adsorption energy affected by combined ligand and coordination effects. (A) Different coordination environments are ranked in increasing order of total coordination number of nearest neighbors, as defined in the text. (B) Frequency distribution of OH* adsorption energy for each coordination environment (left), whose mean values are found to correlate linearly with the total coordination number of nearest neighbors (middle). The coordination types are ordered the same manner in (A) and (B) and the energy values are horizontally aligned in (B). Reproduced with permission[61]. Copyright 2020, Elsevier. MAE: mean absolute error;
HT experimental approach
HT theoretical calculations have shown significant potential in generating a database and predicting novel HEA catalysts with desirable catalytic performance. Most HT theoretical calculation techniques, however, utilize semi-empirical physical and chemical parameters without considering the complexity of such experiments[62]. The synthesis, characterization and performance testing severely affect the reliability of theoretical calculation results. Therefore, the HT experimental approaches should also be explored to assess the structure, property and performance of HEA catalysts, which could further validate the HT theoretical calculation results[63].
For example, Shukla et al. developed a solid-state gradient alloying method for the HT screening of HEA systems, in which a tapered section of a pure alloying element was retrofitted to the base alloy groove via milling and the subsequent friction stir processing of the assembled region achieved a continuous increase in alloy content of the additional element, as shown in Figure 5A-D[64]. This prototype technique was applied to investigate the effect of the gradient variations of Cu on the phases (ε-hcp and γ-fcc) and the mechanical property response of a Fe40Mn20Co20Cr15Si5 HEA. HEA samples with a compositional gradient can also be achieved by diffusion multiple technologies. Zhu et al. applied this technology to the HT synthesis of a Ti-based HEA[65]. By combining the HT diffusion multiple technology and back propagation neural network, a Ti alloy was successfully designed and synthesized, which showed outstanding mechanical properties. HEA products with a continuous composition gradient achieved by HT experiments can be easily characterized by energy-dispersive X-ray spectroscopy (EDS) with scanning electron microscopy (SEM) and are thus suitable for building a database with fewer compositional variables to analyze the composition-activity relationships of HEA catalysts[66].
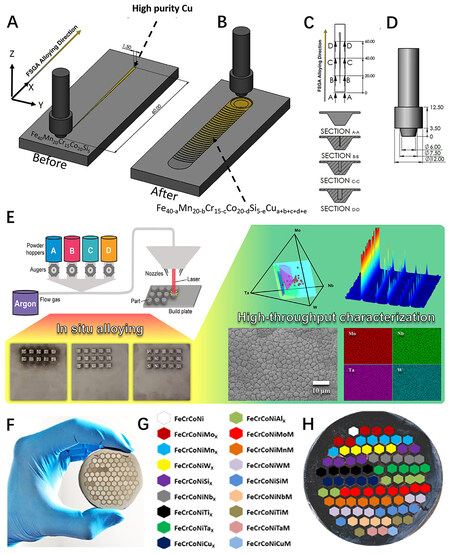
Figure 5. (A) Schematic of friction stir gradient alloying (FSGA) assembly showing tapered Cu section retrofitted in the groove created via CNC (Computer Numerical Control) milling on the base CS-HEA. The total length of the Cu section and groove is 60 mm, with at least 16 mm of base material on either side for tool plunging and retrieval (not drawn to scale). (B) Schematic of processed (alloyed) region after the FSGA process is completed. (C) Location of W-Re tool with respect to the tapered Cu plate during FSGA assembly and (D) design of the tool used in the current study. (E) Schematic of LENS MR-7 system, synthesized Mo-Nb-Ta-W arrays and corresponding HT characterization. (F, G) Design of honeycomb-structured HEAs. (A-D) Reproduced with permission[64]. Copyright 2020, Elsevier. (E) Reproduced with permission[67]. Copyright 2020, Elsevier. (F, G) Reproduced with permission[72]. Copyright 2020, Elsevier. HEA: high-entropy alloy.
Using an Optomec LENS MR-7, the arrays of different HEA compositions were produced by HT additive manufacturing in the form of directed energy deposition[67]. Sample arrays of MoNbTaW HEAs were then characterized by SEM, EDS and X-ray diffraction (XRD). All characterizations were performed
To accelerate the exploration of HEAs with targeted properties or performance, Zhao et al. developed the HT hot-isostatic-pressing-based micro-synthesis approach (HT-HIP-MSA), which can efficiently synthesize and characterize 85 combinatorial alloys in a 13-principal element alloying space, as shown in
Conventional methods for the synthesis of HEAs, such as arc melting, laser cladding, thermal spray, spark plasma sintering and ball milling, are too time-consuming for use in HT techniques for the accelerated discovery of HEAs. To address this issue, our group reported a radio frequency inductively coupled plasma (RF-ICP) method to synthesize HEAs in a rapid and HT fashion[74]. The schematics of the experimental setup and HEA synthesis process using the RF-ICP system are presented in Figure 6A and B. As shown in Figure 6C and D, the time for HEA preparation was within 40 s and ~15 s were needed for cooling the sample. It was found that the porosity of the Cu50Ni50 binary alloy was significantly decreased by increasing the healing time from 9 to 21 s. More importantly, high-purity FeCoNi-based alloys
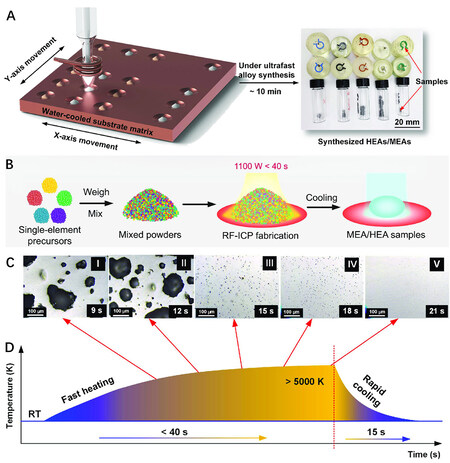
Figure 6. Schematic of (A) HT experimental setup for synthesizing alloys with a large composition space. (B) RF-ICP synthesis of HEAs using mixed pure metal powders. Fast RF-ICP synthesis of alloys using powder mixtures: (C) typical optical microscopy (OM) images showing the evolution of porosity in the Cu50Ni50 alloy under different heating times; (D) temperature profile of RF-ICP synthesis. Reproduced with permission[74]. Copyright 2021, Wiley-VCH. RF-ICP: radio frequency inductively coupled plasma; MEA:
Huang et al. utilized eight or 28 sample holders of one electrode to simultaneously prepare eight or 28 different HEAs, respectively, in one batch of electrolysis under the same conditions[75]. The prepared HEA systems at different locations of the one cathode will not contaminate each other due to the insolubilization of most transition metals and metal oxides in molten salts. Moreover, chemical solution deposition was applied to prepare a library of [Cax(Nb1-yTay)1-x]1-zBizOδ films with a total of 288 compositions[76]. The surface and cross-section microstructures of these designed systems were characterized using field-emission SEM. A HT XRD system was applied to analyze the corresponding crystal structures using synchrotron radiation with a wavelength of 0.8 Å along with a 2D detector (PILATUS) at the SPring-8 facility. This work indicates that implementing HT conductivity measurements and HT XRD allowed us to increase the total experimental throughput for exploring HEA materials.
The HT techniques of computation, synthesis, processing, characterization and data analysis to accelerate the discovery of HEAs are well established. An integrated closed-loop process for HT HEA development, however, has been demonstrated infrequently. Vecchio et al. developed a HT rapid experimental alloy development (HT-READ) methodology, as shown in Figure 7[77]. CALPHAD and ML model-based computational screening provided recommendations for composition selection and sample library design. The designed samples were synthesized, processed, characterized, tested and analyzed in an automated HT fashion. The achieved knowledge was the key to improving the subsequent screening and alloy design. This work indicates that HT theoretical calculations, synthesis and characterization are no longer bottlenecks for the development of HEAs. Note that these methodologies were used to develop HEAs with optimum mechanical properties. It is still challenging to achieve desired catalytic performances in these HT synthesized HEA catalysts, as it is difficult to control the surface properties, morphology and particle size of the synthesized HEAs, which are crucial in the field of catalysis.

Figure 7. Illustration of the steps incorporated into the integrated HT-READ methodology. Clockwise from the top left, computational screening utilizing CALPHAD and the ML model provides recommendations for sample library compositions. The samples are then synthesized, processed, characterized, tested and analyzed in an automated HT fashion. New data are utilized to improve the subsequent screening and design. Reproduced with permission[77]. Copyright 2021, Elsevier. CALPHAD: calculations of phase diagrams; SEM: scanning electron microscopy; XRD: X-ray diffraction; HT-READ: HT rapid experimental alloy development; HT: high-throughput.
To extend the application of HEAs in the catalysis field, Yao et al. reported a HT synthesis technique for the compositional design and rapid thermal-shock treatment of ultrafine HEA nanoclusters (PtPdRhRuIrFeCoNi) with a homogeneous alloy structure, as shown in Figure 8A[78]. In this process, carbon materials with surface defects were used as the supports to ensure the size uniformity for the different composition samples. The HT HEA catalyst systems were then rapidly tested by scanning droplet cell analysis [Figure 8B] for their electrochemical ORR. Their corresponding catalytic performance is displayed in Figure 8C and D, where the two best-performing HEA catalysts were quickly identified. This work indicates that the rapid synthesis and compositional exploration of HEAs by HT techniques are very efficient for exploring HEA catalysts.
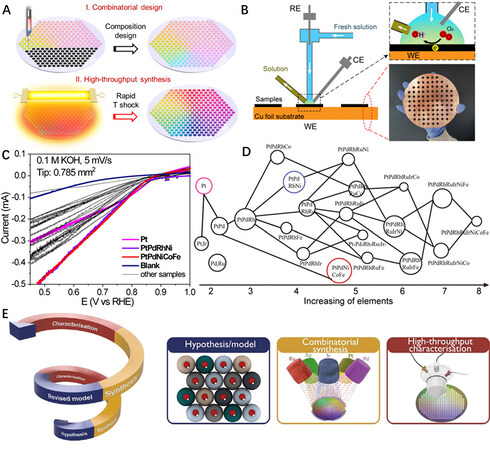
Figure 8. (A) Schematic illustration of combinatorial and HT synthesis of uniform MMNCs. (B) Scanning droplet cell setup and patterned samples on copper substrate. (C) Fast screening of PtPd-based MMNCs for catalytic ORR (22 compositions + one blank, 0.1 M KOH, 5 mV/s scan rate). (D) Compositional designs and their corresponding ORR performance are presented in a neural network diagram. The size of the circles represents the magnitude of the specific current at 0.45 V for ORR. (E) Data-driven discovery cycle combines prediction, combinatorial synthesis and HT characterization. (A-D) Reproduced with permission[78]. Copyright 2019, Science. (E) Reproduced with permission[80]. Copyright 2021, Wiley-VCH. CE: counter electrode; RE: reference electrode; WE: working electrode.
The co-sputtering method has shown significant potential for the HT synthesis of HEAs due to the small grain size, few defects and low contamination in film samples[74,79]. More importantly, this HT co-sputtering approach has been utilized successfully to synthesize HEA catalysts and build a library. The Ludwig group demonstrated a closed-loop, data-driven HT experimentation technique that iteratively combines DFT calculations, the combinatorial synthesis of material libraries and HT characterization [Figure 8E][80]. In this work, three Ag-Ir-Pd-Pt-Ru material libraries with large compositional spaces, centered around the predicted compositions, were prepared by combinatorial co-sputtering of the five elemental targets. The refined model, with the input derived from HT characterization data sets, could predict the activity of the exemplary Ag-Ir-Pd-Pt-Ru model system and further identify optimal HEA catalysts in an unprecedented manner. Furthermore, they extended this strategy to the HEA systems of Rh-Ir-Pd-Pt-Ru to unravel the composition-activity-stability relations in HEA electrocatalysts[81].
ML MODELS FOR HEA CATALYSTS
ML is a powerful tool for accelerating catalyst discovery, as it can be used to build models with high accuracy, predict the catalytic performance of unknown catalysts and understand the
ML models of ammonia decomposition and synthesis on HEA catalysts
Based on HT calculations, a database with 1911 configurations was generated by Saidi et al., where the adsorption energies of N* were within the range of -2.4-1.2 eV on CoMoFeNiCu HEAs[56]. The large number of adsorption sites with different chemical environments results in the large variance in the adsorption energies of N*. This means that such a wide variation requires training the data set with a wide range of different systems, thereby increasing the training time for accurate ML. To consider the symmetry of active sites, the data set was extended by 10%-25%. Taking the HCP-hollow site as an example, little variation in the adsorption energy can be observed when changing the arrangement of three
ML models of ORR on HEA catalysts
The Rossmeisl group performed HT DFT calculations for the adsorption energies of OH* and O* on 871 and 998 different 2 × 2 unit cells of IrPdPtRhRu HEAs, respectively, as summarized in Figure 9A and B[60]. According to these data, the authors trained a ML model using the ordinary least squares algorithm to predict the full span of available adsorption energies on the HEA (111) surface. As shown in
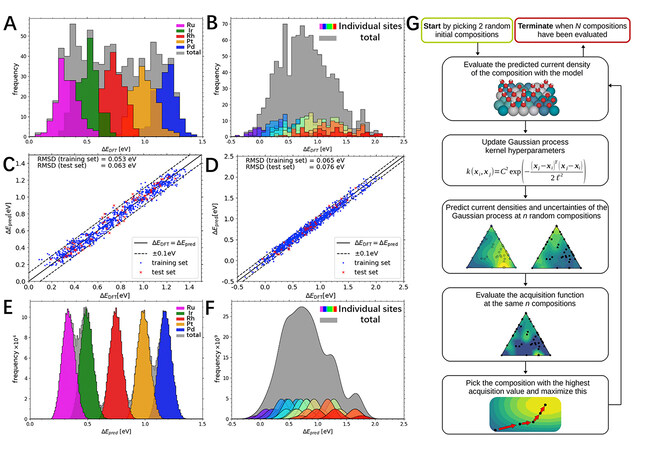
Figure 9. (A) OH* adsorption energies for 871 2 × 2 periodic unit cells. (B) O* adsorption energies for 998 2 × 2 periodic unit cells. (C) OH* adsorption. The linear model was trained on 871 symmetric 2 × 2 unit cells (blue dots) and tested on 76 asymmetric 3 × 4 unit cells (red crosses). The linear model used 15 parameters. (D) O* adsorption. The linear model was trained on 998 symmetric 2 × 2 unit cells (blue dots) and tested on 36 asymmetric 3 × 4 unit cells (red crosses). The linear model used 55 parameters. The dashed lines span the region ± 0.1 eV, where most of the data were seen to be contained. (E) OH* adsorption. Each color represents an individual on-top binding site as in (A). (F) O* adsorption. Each color represents an individual FCC hollow binding site, as shown in (B). (G) Workflow of Bayesian optimization algorithm. The algorithm was terminated after n = 150 samples to ensure sufficient evaluations for gauging the deviation in the number of samples needed for the discovery of the optimal compositions. For evaluation of the acquisition function, n = 1000 random compositions were sampled. (A-F) Reproduced with permission[60]. Copyright 2019, Elsevier. (G) Reproduced with permission[84]. Copyright 2021, Wiley-VCH. FCC: face-centered cubic.
ML models of CO2RR on HEA catalysts
The ever-increasing demand for global energy and the need to replace CO2-emitting fossil fuels with renewable sources have driven interest in energy conversion and storage. In particular, the electrochemical reduction of CO2 to chemical feedstocks is a hot topic due to its high correlation with both CO2 removal and renewable energy generation. To accelerate catalyst discovery for the CO2RR, Zhong et al. developed a
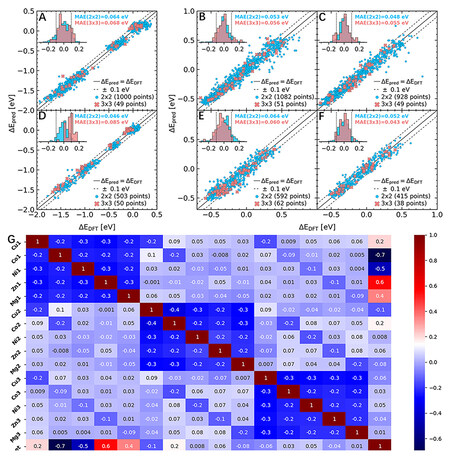
Figure 10. Plots showing GPR-predicted (ΔEpred) versus DFT-calculated (ΔEDFT) adsorption energies for (A-C) CoCuGaNiZn and (D-F) AgAuCuPdPt for (A, D) on-top CO, (B, E) FCC-hollow H and (C, F) HCP-hollow H. Blue and red indicate data for 2 × 2 and 3 × 3 atom slabs. MAEs are calculated as a fivefold cross-validation prediction error for the 2 × 2 and 3 × 3 slabs as the prediction error when training on the set of all 2 × 2 slabs. The insets show the distribution of the prediction errors in eV defined as ΔEpred-ΔEDFT. (G) correlation matrix of all the input features and output (target) adsorption energy for CO*, where 1, 2and 3 represent the first, second and third regions of the microstructure, respectively. (A-F) Reproduced with permission[86]. Copyright 2020, American Chemical Society. (G) Reproduced with permission[53]. Copyright 2021, American Chemical Society. HCP: hexagonal-closed packed; FCC: face-centered cubic; MAE: mean absolute error.
Descriptors in ML models of HEA catalysts
The key to constructing a ML model is designing effective descriptors, which is more important for HEA catalysts due to the complex active sites. The appropriate descriptors as input features for a ML model should be achieved directly from databases or by the simplest DFT calculations and include sufficient information on surface active sites. Some approaches, such as coordination atom fingerprints (CAFs), Coulomb matrices, the spectrum of London and Axilrod-Teller-Muto, elemental properties and SLATM (EP & SLATM), smooth overlap of atomic positions, Voronoi connectivity-based crystal graph, labeled site crystal graph (LSCG) and FCHL19, have recently been reported[87-94]. Li et al. applied elemental groups and periods (GP) to replace atomic numbers in the FCHL19, LSCG, Atomic Number and Coordination Number (ANCN) and CAF representations to achieve an effective improvement for predicting adsorption energies on alloys[95]. This strategy effectively enables ML models to learn from the periodic table. An improvement is achieved up to ~0.2 eV in adsorption energy MAE, compared to those obtained using ANCN, CAF, FCHL19 and LSCG. In particular, for the GP-LSCG representation, the MAE is 0.05 eV (near chemical accuracy) in predicting hydrogen adsorption and ~0.1 eV for other strong binding adsorbates (C*, N*, O* and S*). Although this work mainly focuses on bimetallic alloy systems, it has the potential to be extended to HEA catalysts, which has been verified by another research group, who proposed a transferable ML model by considering the intrinsic properties of substrates and adsorbates[96]. Simply training the properties of transition metals could predict the adsorption energies of single atom alloys, AB intermetallics and HEAs through understanding the relation between some descriptors (surface atom valence, electronegativity, coordination and adsorbate valence) and the adsorption energy. This transferable scheme could achieve new insights into the adsorption mechanism on HEA surfaces and the rapid design of HEA catalysts.
Although the current applications of ML in the field of HEA catalysts are limited to the reactions discussed above, some HEAs have shown excellent catalytic performance for some other reactions[18,19,97]. For instance, Wang et al. developed a new class of structurally ordered PtRhFeNiCu HEAs as electrocatalysts for the ethanol oxidation reaction[98]. Feng et al. synthesized ultrasmall HEA nanoparticles with an average diameter of 1.68 nm by a suitable and scalable synthetic strategy, which achieved an ultrahigh mass activity of
CHALLENGES
HEA catalyst research is in its infancy and some open questions for synthetic methods, catalytic reactions and mechanistic understandings should be addressed. As the number of components increases, the active centers of HEAs become much more complex compared to traditional alloy systems. Thus, the key study in the research of HEA catalysts is the identification of active centers. The development of HT techniques and ML models is an essential part of the accelerated research of HEA catalysts with a huge compositional space, as illustrated in Figure 11[25]. However, more challenges are still unresolved for the development of HT techniques and ML models for HEA catalysts, as shown below:
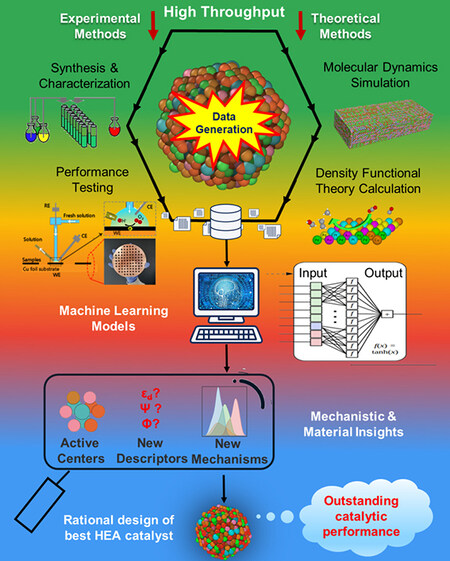
Figure 11. New research strategy for HEA catalysts. HT experimental and theoretical methods are used to generate comprehensive databases of HEA catalysts. Highly transferable and accurate ML models are explored to analyze databases and predict optimal HEA catalysts. New insights into active centers and new catalytic mechanisms and descriptors are expected to be developed on the basis of HT techniques and ML models. Finally, high-performance HEA catalysts will be rationally designed, promoting the development of catalysis. Reproduced with permission[25]. Copyright 2022, Elsevier. CE: counter electrode; RE: reference electrode; WE: working electrode; HEA: high-entropy alloy; HT: high-throughput; ML: machine learning.
(1) The morphology and particle size of HEA catalysts should have obvious influences on the catalytic performance; however, they are difficult to control flexibly by current HT synthesis techniques;
(2) Though many HT synthesis techniques have been successful, the synthesized HEAs are not easy to further test for catalytic performance;
(3) Current HT calculations are mainly focused on the adsorption of some important intermediates on HEA catalysts, rather than the most intuitive catalytic performance. This strategy effectively reduces the cost of computation; however, it only works when the BEP and scaling relationships between the adsorption energies of the relevant intermediates are still valid on the surface of the HEA catalysts;
(4) Although ML has a powerful prediction ability, the accuracy depends on the sufficiency of training data. The existing databases contain many valuable material data; unfortunately, there are still more data in the published literature that cannot be entered into databases and shared. Therefore, a more comprehensive and generic material information standard should be established to achieve data sharing among databases and to reduce obstacles in data acquisition;
(5) The key challenge in ML is the exploration of suitable, simple and general descriptors to accurately describe HEA catalysts, which are required to reasonably design catalysts and efficiently screen candidates;
(6) The prediction ability of ML models based on HT techniques has been proven to be powerful. Another important milestone is to uncover the structure-property-performance relationships of HEA catalysts and explore new mechanisms in the catalysis field, which deserves more attention in future research.
PERSPECTIVES
To address the above challenges, more HT synthesis strategies, as well as HT techniques for characterizing and testing catalytic performances, should be explored to build databases for HEA catalysts. The achieved databases should be more comprehensive, realistic, reliable and universal. In addition to the composition of HEA catalysts, we should also pay attention to other parameters (such as morphology, size, specific surface area and reaction environments), which are directly related to catalytic performances. For HT calculations, a more comprehensive list of catalytic performance parameters should be considered to evaluate the catalytic performance of HEA catalysts, rather than only calculating the adsorption energy of some important intermediates. Because both the BEP relationship and scaling relationship of adsorption energies are currently controversial phenomena for HEA catalysts, which need to be further verified. With their complex surface active sites, HEA catalysts have shown excellent catalytic activity for various catalytic reactions. The selectivity of HEA catalysts, however, is also a concern as these complex active centers might catalyze multiple reactions, which is another challenge for the development of HEA catalysts. To make full use of the existing data (both experimental and calculation data) in published studies and databases, and avoid the batch effect, workflows with natural language processing techniques should be explored to achieve effective communication between humans and computers with human languages and integrate these useful data into a comprehensive database. Finally, more effective descriptors should be proposed, as the accuracy and universality of the designed ML models are determined by the descriptors adopted. An effective descriptor can accelerate our understanding of HEA catalysts and help us to discover new catalytic mechanisms, thus promoting the development of HEA catalysts. We hope that this review will help researchers to better understand the significance of HT techniques and ML models for the development of HEA catalysts.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception and design of this review, writing and editing: Chen L, Singh CV, Zou Y
Made substantial contributions to collation of literatures, figures preparation, and writing: Chen L, Chen Z, Singh CV, Zou Y
Performed data analysis, discussion and writing review: Chen L, Chen Z, Yao X, Su B, Chen W, Pang X, Kim KS, Singh CV, Zou Y
Performed data acquisition and interpretation: Chen L, Zou Y
Availability of data and materialsNot applicable.
Financial support and sponsorshipCVS acknowledges financial support from the Nature Science and Engineer Research Council of Canada (NSERC), Hart Professorship, and the University of Toronto. YZ acknowledges the financial support from Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant # RGPIN-2018-05731), LXC, KSK and YZ acknowledge the financial support from Collaboration Centre in Green Energy Materials (CC-GEM- #2020-0424). XP acknowledge the financial support from the Office of Energy Research and Development (OERD), Natural Resources Canada.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Meirer F, Weckhuysen BM. Spatial and temporal exploration of heterogeneous catalysts with synchrotron radiation. Nat Rev Mater 2018;3:324-40.
2. Toniato A, Vaucher AC, Laino T. Grand challenges on accelerating discovery in catalysis. Catal Today 2022;387:140-2.
4. Seh ZW, Kibsgaard J, Dickens CF, Chorkendorff I, Nørskov JK, Jaramillo TF. Combining theory and experiment in electrocatalysis: insights into materials design. Science 2017;355:eaad4998.
5. Guan Q, Zhu C, Lin Y, et al. Bimetallic monolayer catalyst breaks the activity-selectivity trade-off on metal particle size for efficient chemoselective hydrogenations. Nat Catal 2021;4:840-9.
6. O’connor NJ, Jonayat ASM, Janik MJ, Senftle TP. Interaction trends between single metal atoms and oxide supports identified with density functional theory and statistical learning. Nat Catal 2018;1:531-9.
7. Liu L, Corma A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem Rev 2018;118:4981-5079.
8. Cui X, Li W, Ryabchuk P, Junge K, Beller M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat Catal 2018;1:385-97.
9. Wang ZL, Yan JM, Ping Y, Wang HL, Zheng WT, Jiang Q. An efficient CoAuPd/C catalyst for hydrogen generation from formic acid at room temperature. Angew Chem Int Ed Engl 2013;52:4406-9.
10. Lang X, Han G, Xiao B, et al. Mesostructured intermetallic compounds of platinum and non-transition metals for enhanced electrocatalysis of oxygen reduction reaction. Adv Funct Mater 2015;25:230-7.
11. Qin Y, Zhang W, Guo K, et al. Fine-tuning intrinsic strain in penta-twinned Pt-Cu-Mn nanoframes boosts oxygen reduction catalysis. Adv Funct Mater 2020;30:1910107.
12. Yao R, Zhou Y, Shi H, et al. Nanoporous surface high-entropy alloys as highly efficient multisite electrocatalysts for nonacidic hydrogen evolution reaction. Adv Funct Mater 2021;31:2009613.
13. Yao Y, Dong Q, Brozena A, et al. High-entropy nanoparticles: synthesis-structure-property relationships and data-driven discovery. Science 2022;376:eabn3103.
14. Cantor B, Chang I, Knight P, Vincent A. Microstructural development in equiatomic multicomponent alloys. Mater Sci Eng A 2004;375-377:213-8.
15. Yeh J, Chen S, Lin S, et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv Eng Mater 2004;6:299-303.
16. Cheng C, Zhang X, Haché MJR, Zou Y. Phase transition and nanomechanical properties of refractory high-entropy alloy thin films: effects of co-sputtering Mo and W on a TiZrHfNbTa system. Nanoscale 2022;14:7561-8.
17. Cheng C, Zhang X, Haché MJR, Zou Y. Magnetron co-sputtering synthesis and nanoindentation studies of nanocrystalline (TiZrHf)x(NbTa)1-x high-entropy alloy thin films. Nano Res 2022;15:4873-9.
18. Xin Y, Li S, Qian Y, et al. High-entropy alloys as a platform for catalysis: progress, challenges, and opportunities. ACS Catal 2020;10:11280-306.
19. Ma Y, Ma Y, Wang Q, et al. High-entropy energy materials: challenges and new opportunities. Energy Environ Sci 2021;14:2883-905.
20. Xie P, Yao Y, Huang Z, et al. Highly efficient decomposition of ammonia using high-entropy alloy catalysts. Nat Commun 2019;10:4011.
21. Mori K, Hashimoto N, Kamiuchi N, Yoshida H, Kobayashi H, Yamashita H. Hydrogen spillover-driven synthesis of high-entropy alloy nanoparticles as a robust catalyst for CO2 hydrogenation. Nat Commun 2021;12:3884.
22. Wang D, Chen Z, Huang Y, et al. Tailoring lattice strain in ultra-fine high-entropy alloys for active and stable methanol oxidation. Sci China Mater 2021;64:2454-66.
23. Wu D, Kusada K, Nanba Y, et al. Noble-metal high-entropy-alloy nanoparticles: atomic-level insight into the electronic structure. J Am Chem Soc 2022;144:3365-9.
24. Li H, Han Y, Zhao H, et al. Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis. Nat Commun 2020;11:5437.
25. Chen ZW, Chen L, Gariepy Z, Yao X, Singh CV. High-throughput and machine-learning accelerated design of high entropy alloy catalysts. Trends Chem 2022;4:577-9.
27. Wan X, Zhang Z, Niu H, et al. Machine-learning-accelerated catalytic activity predictions of transition metal phthalocyanine dual-metal-site catalysts for CO2 reduction. J Phys Chem Lett 2021;12:6111-8.
28. Chen ZW, Lu Z, Chen LX, Jiang M, Chen D, Singh CV. Machine-learning-accelerated discovery of single-atom catalysts based on bidirectional activation mechanism. Chem Catal 2021;1:183-95.
29. Zafari M, Kumar D, Umer M, Kim KS. Machine learning-based high throughput screening for nitrogen fixation on boron-doped single atom catalysts. J Mater Chem A 2020;8:5209-16.
30. Deng C, Su Y, Li F, Shen W, Chen Z, Tang Q. Understanding activity origin for the oxygen reduction reaction on bi-atom catalysts by DFT studies and machine-learning. J Mater Chem A 2020;8:24563-71.
31. Wan X, Zhang Z, Yu W, Niu H, Wang X, Guo Y. Machine-learning-assisted discovery of highly efficient high-entropy alloy catalysts for the oxygen reduction reaction. Patterns (N Y) 2022;3:100553.
32. Roy D, Mandal SC, Pathak B. Machine learning assisted exploration of high entropy alloy-based catalysts for selective CO2 reduction to methanol. J Phys Chem Lett 2022;13:5991-6002.
33. Nandy A, Duan C, Taylor MG, Liu F, Steeves AH, Kulik HJ. Computational discovery of transition-metal complexes: from high-throughput screening to machine learning. Chem Rev 2021;121:9927-10000.
34. Rodríguez-Martínez X, Pascual-San-José E, Campoy-Quiles M. Accelerating organic solar cell material’s discovery: high-throughput screening and big data. Energy Environ Sci 2021;14:3301-22.
35. Conway PL, Klaver T, Steggo J, Ghassemali E. High entropy alloys towards industrial applications: high-throughput screening and experimental investigation. Mater Sci Eng A 2022;830:142297.
36. Miracle D, Majumdar B, Wertz K, Gorsse S. New strategies and tests to accelerate discovery and development of multi-principal element structural alloys. Scr Mater 2017;127:195-200.
37. Mannodi-kanakkithodi A, Chan MK. Computational data-driven materials discovery. Trends Chem 2021;3:79-82.
38. Huang E, Lee W, Singh SS, et al. Machine-learning and high-throughput studies for high-entropy materials. Mater Sci Eng R Rep 2022;147:100645.
39. Lederer Y, Toher C, Vecchio KS, Curtarolo S. The search for high entropy alloys: a high-throughput ab-initio approach. Acta Mater 2018;159:364-83.
40. Ångqvist M, Muñoz WA, Rahm JM, et al. ICET - a python library for constructing and sampling alloy cluster expansions. Adv Theory Simul 2019;2:1900015.
41. van de Walle A, Tiwary P, de Jong M, et al. Efficient stochastic generation of special quasirandom structures. Calphad 2013;42:13-8.
42. de Walle A, Asta M, Ceder G. The alloy theoretic automated toolkit: a user guide. Calphad 2002;26:539-53.
43. Okhotnikov K, Charpentier T, Cadars S. Supercell program: a combinatorial structure-generation approach for the local-level modeling of atomic substitutions and partial occupancies in crystals. J Cheminform 2016;8:17.
44. Zunger A, Wei S, Ferreira LG, Bernard JE. Special quasirandom structures. Phys Rev Lett 1990;65:353-6.
45. Feugmo CG, Ryczko K, Anand A, Singh CV, Tamblyn I. Neural evolution structure generation: high entropy alloys. J Chem Phys 2021;155:044102.
46. Li B, Li X, Gao W, Jiang Q. An effective scheme to determine surface energy and its relation with adsorption energy. Acta Mater 2021;212:116895.
47. Sharma M, Jang J, Shin DY, et al. Work function-tailored graphene
48. Duong T, Wang Y, Yan X, Couet A, Chaudhuri S. A first-principles-based approach to the high-throughput screening of corrosion-resistant high entropy alloys. arXiv preprint arXiv 2021;2104:10590.
49. Abild-Pedersen F, Greeley J, Studt F, et al. Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces. Phys Rev Lett 2007;99:016105.
50. Nørskov J, Bligaard T, Logadottir A, et al. Universality in heterogeneous catalysis. J Catal 2002;209:275-8.
51. Kuhl KP, Cave ER, Abram DN, Jaramillo TF. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ Sci 2012;5:7050.
52. Chen ZW, Gao W, Zheng WT, Jiang Q. Steric hindrance in sulfur vacancy of monolayer MoS2 boosts electrochemical reduction of carbon monoxide to methane. ChemSusChem 2018;11:1455-9.
53. Roy D, Mandal SC, Pathak B. Machine learning-driven high-throughput screening of alloy-based catalysts for selective CO2 hydrogenation to methanol. ACS Appl Mater Inter 2021;13:56151-63.
54. Singh AR, Rohr BA, Schwalbe JA, et al. Electrochemical ammonia synthesis - the selectivity challenge. ACS Catal 2017;7:706-9.
55. Chen Z, Lang XY, Jiang Q. Discovery of cobweb-like MoC6 and its application for nitrogen fixation. J Mater Chem A 2018;6:9623-8.
56. Saidi WA, Shadid W, Veser G. Optimization of high-entropy alloy catalyst for ammonia decomposition and ammonia synthesis. J Phys Chem Lett 2021;12:5185-92.
57. Montoya JH, Tsai C, Vojvodic A, Nørskov JK. The challenge of electrochemical ammonia synthesis: a new perspective on the role of nitrogen scaling relations. ChemSusChem 2015;8:2180-6.
58. Skúlason E, Bligaard T, Gudmundsdóttir S, et al. A theoretical evaluation of possible transition metal electro-catalysts for N2reduction. Phys Chem Chem Phys 2012;14:1235-45.
59. Nørskov JK, Rossmeisl J, Logadottir A, et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J Phys Chem B 2004;108:17886-92.
60. Batchelor TA, Pedersen JK, Winther SH, Castelli IE, Jacobsen KW, Rossmeisl J. High-entropy alloys as a discovery platform for electrocatalysis. Joule 2019;3:834-45.
61. Lu Z, Chen ZW, Singh CV. Neural network-assisted development of high-entropy alloy catalysts: decoupling ligand and coordination effects. Matter 2020;3:1318-33.
62. Saidi WA. Emergence of local scaling relations in adsorption energies on high-entropy alloys. NPJ Comput Mater 2022:8.
63. McCullough K, Williams T, Mingle K, Jamshidi P, Lauterbach J. High-throughput experimentation meets artificial intelligence: a new pathway to catalyst discovery. Phys Chem Chem Phys 2020;22:11174-96.
64. Shukla S, Wang T, Frank M, et al. Friction stir gradient alloying: a novel solid-state high throughput screening technique for high entropy alloys. Mater Today Commun 2020;23:100869.
65. Zhu C, Li C, Wu D, et al. A titanium alloys design method based on high-throughput experiments and machine learning. J Mater Res Technol 2021;11:2336-53.
66. Coury FG, Wilson P, Clarke KD, Kaufman MJ, Clarke AJ. High-throughput solid solution strengthening characterization in high entropy alloys. Acta Mater 2019;167:1-11.
67. Moorehead M, Bertsch K, Niezgoda M, et al. High-throughput synthesis of Mo-Nb-Ta-W high-entropy alloys via additive manufacturing. Mater Des 2020;187:108358.
68. Pegues JW, Melia MA, Puckett R, Whetten SR, Argibay N, Kustas AB. Exploring additive manufacturing as a high-throughput screening tool for multiphase high entropy alloys. Addit Manuf 2021;37:101598.
69. Dobbelstein H, George EP, Gurevich EL, Kostka A, Ostendorf A, Laplanche G. Laser metal deposition of refractory high-entropy alloys for high-throughput synthesis and structure-property characterization. Int J Extrem Manuf 2021;3:015201.
70. Li M, Gazquez J, Borisevich A, Mishra R, Flores KM. Evaluation of microstructure and mechanical property variations in AlxCoCrFeNi high entropy alloys produced by a high-throughput laser deposition method. Intermetallics 2018;95:110-8.
71. Li M, Flores KM. Laser processing as a high-throughput method to investigate microstructure-processing-property relationships in multiprincipal element alloys. J Alloys Compd 2020;825:154025.
72. Zhao L, Jiang L, Yang L, et al. High throughput synthesis enabled exploration of CoCrFeNi-based high entropy alloys. J Mater Sci Technol 2022;110:269-82.
73. Xu Y, Bu Y, Liu J, Wang H. In-situ high throughput synthesis of high-entropy alloys. Scr Mater 2019;160:44-7.
74. Zhu B, Alavi S, Cheng C, et al. Fast and High-throughput synthesis of medium- and high-entropy alloys using radio frequency inductively coupled plasma. Adv Eng Mater 2021;23:2001116.
75. Huang J, Shi H, Ma Y, Yin H, Wang D. A combinatorial electrode for high-throughput, high-entropy alloy screening. ChemElectroChem 2021;8:4573-9.
76. Matsubara M, Suzumura A, Ohba N, Asahi R. Identifying superionic conductors by materials informatics and high-throughput synthesis. Commun Mater 2020:1.
77. Vecchio KS, Dippo OF, Kaufmann KR, Liu X. High-throughput rapid experimental alloy development (HT-READ). Acta Mater 2021;221:117352.
78. Yao Y, Huang Z, Li T, et al. High-throughput, combinatorial synthesis of multimetallic nanoclusters. Proc Natl Acad Sci U S A 2020;117:6316-22.
79. Shi Y, Yang B, Rack PD, Guo S, Liaw PK, Zhao Y. High-throughput synthesis and corrosion behavior of sputter-deposited nanocrystalline Alx(CoCrFeNi)100-x combinatorial high-entropy alloys. Mater Des 2020;195:109018.
80. Batchelor TAA, Löffler T, Xiao B, et al. Complex-solid-solution electrocatalyst discovery by computational prediction and high-throughput experimentation*. Angew Chem Int Ed Engl 2021;60:6932-7.
81. Banko L, Krysiak OA, Pedersen JK, et al. Unravelling composition-activity-stability trends in high entropy alloy electrocatalysts by using a data-guided combinatorial synthesis strategy and computational modeling. Adv Energy Mater 2022;12:2103312.
82. Yang Z, Gao W. Applications of machine learning in alloy catalysts: rational selection and future development of descriptors. Adv Sci (Weinh) 2022;9:e2106043.
83. Gao W, Chen Y, Li B, Liu SP, Liu X, Jiang Q. Determining the adsorption energies of small molecules with the intrinsic properties of adsorbates and substrates. Nat Commun 2020;11:1196.
84. Pedersen JK, Clausen CM, Krysiak OA, et al. Bayesian optimization of high-entropy alloy compositions for electrocatalytic oxygen reduction*. Angew Chem Int Ed Engl 2021;60:24144-52.
85. Zhong M, Tran K, Min Y, et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 2020;581:178-83.
86. Pedersen JK, Batchelor TAA, Bagger A, Rossmeisl J. High-entropy alloys as catalysts for the CO2 and CO reduction reactions. ACS Catal 2020;10:2169-76.
87. Tran K, Ulissi ZW. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat Catal 2018;1:696-703.
88. Li Z, Ma X, Xin H. Feature engineering of machine-learning chemisorption models for catalyst design. Catal Today 2017;280:232-8.
89. Huang B, von Lilienfeld OA. Quantum machine learning using atom-in-molecule-based fragments selected on the fly. Nat Chem 2020;12:945-51.
90. Li X, Chiong R, Hu Z, Cornforth D, Page AJ. Improved representations of heterogeneous carbon reforming catalysis using machine learning. J Chem Theory Comput 2019;15:6882-94.
92. Back S, Yoon J, Tian N, Zhong W, Tran K, Ulissi ZW. Convolutional neural network of atomic surface structures to predict binding energies for high-throughput screening of catalysts. J Phys Chem Lett 2019;10:4401-8.
93. Gu GH, Noh J, Kim S, Back S, Ulissi Z, Jung Y. Practical deep-learning representation for fast heterogeneous catalyst screening. J Phys Chem Lett 2020;11:3185-91.
94. Christensen AS, Bratholm LA, Faber FA, Anatole von Lilienfeld O. FCHL revisited: faster and more accurate quantum machine learning. J Chem Phys 2020;152:044107.
95. Li X, Chiong R, Page AJ. Group and period-based representations for improved machine learning prediction of heterogeneous alloy catalysts. J Phys Chem Lett 2021;12:5156-62.
96. Li X, Li B, Yang Z, Chen Z, Gao W, Jiang Q. A transferable machine-learning scheme from pure metals to alloys for predicting adsorption energies. J Mater Chem A 2022;10:872-80.
97. Fu M, Ma X, Zhao K, Li X, Su D. High-entropy materials for energy-related applications. iScience 2021;24:102177.
98. Wang D, Chen Z, Wu Y, et al. Structurally ordered high-entropy intermetallic nanoparticles with enhanced C-C bond cleavage for ethanol oxidation. SmartMat 2022; doi: 10.1002/smm2.1117.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chen L, Chen Z, Yao X, Su B, Chen W, Pang X, Kim KS, Singh CV, Zou Y. High-entropy alloy catalysts: high-throughput and machine learning-driven design. J Mater Inf 2022;2:19. http://dx.doi.org/10.20517/jmi.2022.23
AMA Style
Chen L, Chen Z, Yao X, Su B, Chen W, Pang X, Kim KS, Singh CV, Zou Y. High-entropy alloy catalysts: high-throughput and machine learning-driven design. Journal of Materials Informatics. 2022; 2(4): 19. http://dx.doi.org/10.20517/jmi.2022.23
Chicago/Turabian Style
Chen, Lixin, Zhiwen Chen, Xue Yao, Baoxian Su, Weijian Chen, Xin Pang, Keun-Su Kim, Chandra Veer Singh, Yu Zou. 2022. "High-entropy alloy catalysts: high-throughput and machine learning-driven design" Journal of Materials Informatics. 2, no.4: 19. http://dx.doi.org/10.20517/jmi.2022.23
ACS Style
Chen, L.; Chen Z.; Yao X.; Su B.; Chen W.; Pang X.; Kim K.S.; Singh CV.; Zou Y. High-entropy alloy catalysts: high-throughput and machine learning-driven design. J. Mater. Inf. 2022, 2, 19. http://dx.doi.org/10.20517/jmi.2022.23
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 34 clicks
Cite This Article 34 clicks

 Like This Article 14
likes
Like This Article 14
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.